Achtergronden
Dit hoofdstuk geeft u achtergrondinformatie bij het 22q11.2DS. Hoe vaak komt het voor? Hoe ontstaat het? En wat is bekend over het toekomstperspectief: het beloop, de prognose en de levensverwachting?
Er wordt geschat dat 22q11.2DS voor komt bij circa 1:2000 tot 4000 levendgeborenen. Het is lastig een exacte schatting te maken. Het is het meest voorkomende autosomale microdeletiesyndroom.
In Nederland worden er per jaar naar schatting ongeveer 50 kinderen met 22q11.2DS geboren. Er worden ongeveer evenveel jongens als meisjes geboren en het risico is even groot bij alle verschillende etniciteiten. Bij een groot deel van de mensen met 22q11.2DS wordt de diagnose echter pas later in het leven gesteld (veelal op de kinderleeftijd).
Een huisarts met een normpraktijk ziet gemiddeld hoogstens één persoon met 22q11.2DS.
Het 22q11.2DS wordt veroorzaakt door een ontbrekend stukje (deletie) van de lange arm van chromosoom 22.
- De deletie kan wisselend van grootte zijn.
- Ongeveer 85% van de mensen met 22q11.2DS heeft dezelfde 2.54 Mb (A-D) deletie (voorheen beschreven als 3 Mb deletie).
ongeveer 5% van de mensen heeft een kleinere 1.5Mb (A-B) deletie. Er zijn geen duidelijke klinische verschillen bij personen met de kleinere deletie in vergelijking met personen met een 2.54 Mb deletie.
In een klein deel van de mensen komen andere/atypische deleties voor (b.v. D-E). Bij deze mensen wordt een ander klinisch beeld gezien. - Bij ongeveer 90% van de mensen met 22q11.2DS is de deletie de novo ontstaan. Bij de overige 10% is de aandoening overgedragen door één van de ouders.
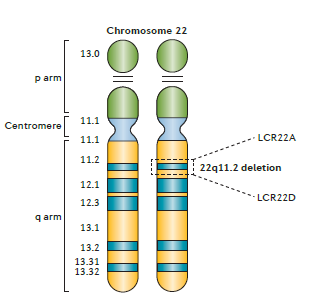
De 22q11.2 regio bevat verschillende blokken van zogenoemde low-copy repeats (LCRs). Dit zijn gebieden van segmentale duplicatie, stukken in een chromosoom, die een aantal keren voorkomen vlak achter elkaar en waarvan de DNA-code veel op elkaar lijkt. Als gevolg van malalignment (ongelijke uitwisseling tijdens de meiose) of non-allelische recombinatie (dezelfde stukken DNA staan niet tegenover elkaar, de chromosomenparen staan scheef) tijdens de eerste meiose kunnen dan deleties en duplicaties ontstaan. De meest voorkomende 22q11.2 deletie (A-D) bevat circa 90 genen, waarvan ongeveer de helft eiwit-coderend.
Uitleg van bovenstaande: Bij een specifieke celdeling bij de de aanmaak van zaad en eicel (Meiose) vindt er een uitwisseling plaats van chromosoomstukken. Hierbij ruilen dezelfde stukken DNA van beide chromosomen van een chromosoompaar van plek – dus verhuizen van het ene chromosoom naar het andere. Hiervoor moeten chromosomen tegenover elkaar gaan liggen. De LCR’s fungeren als herkenningspunten. Als dit niet goed gebeurt krijg je als het ware een jas die verkeerd is dichtgeknoopt (De knopen zijn dan de LCR regio’s). In dat geval kan de uitwisseling van het stuk DNA fout gaan. En kunnen er foute geslachtscellen ontstaan. Een met een chromosoom dat een stuk DNA mist en een deletie 22q11.2 bevat. En de andere geslachtscel met het andere chromosoom dus dat stuk juist te veel (duplicatie 22q11.2).
De genetische diagnose kan op verschillende manieren worden gesteld. Lees meer over genetisch onderzoek in het hoofdstuk Diagnostiek.
Wist u dat…...... er ook een (micro) duplicatie 22q11.2 voorkomt? Hierbij is een stukje op chromosoom 22 te veel aanwezig (22q11.2 duplicatie syndroom.) Alhoewel het dus om een andere genetische aandoening gaat, is er een overlap in het klinisch beeld met 22q11.2DS.
Omdat er nog te weinig specifieke kennis over 22q11.2 duplicatie syndroom is, worden in de praktijk meestal dezelfde preventieve onderzoeken gedaan als bij 22q11.2DS. Het 22q11.2 duplicatie syndroom wordt hier nu verder niet besproken.
Beloop
Het beloop tussen verschillende personen met 22q11.2DS is zeer variabel. Veel kinderen met 22q11.2DS kampen met verschillende lichamelijke problemen, waaronder aangeboren aandoeningen van het hart en gehemelte, immuunstoornissen met recidiverende ontstekingen en voedingsproblemen als gevolg van onder meer gehemelteafwijkingen. Ontwikkelingsachterstand, psychiatrische aandoeningen en ander consequenties van het hebben van 22q11.2DS kunnen verstrekkende effecten hebben op het dagelijks functioneren en de kwaliteit van leven van de persoon met 22q11.2DS, maar ook van diens familie.
De kwaliteit van leven van de persoon met 22q11.2DS en zijn/haar omgeving hangt o.a. af van problemen in het cognitieve, sociale en emotionele domein. Bovendien zijn het aantal ziekenhuisopnames, meer ernstige immunologische problemen en de complexiteit van het aantal aangedane orgaansystemen van invloed. Vlotte signalering van doorgaans goed behandelbare aandoeningen (waaronder psychiatrische stoornissen) en snelle interventies, goede zorg en ondersteuning kunnen helpen om de kwaliteit van leven aanzienlijk te verbeteren. Vaak wordt ook geadviseerd maatregelen te nemen die de kans op “overvragen” verkleinen, zoals overgang naar speciaal (basis / voortgezet) onderwijs.
Prognose en levensverwachting
Dankzij de huidige goede medische zorg, is de gemiddelde levensverwachting bij de meeste mensen met 22q11.2DS zoals bij de algemene populatie. Specifiek na de geboorte kan er wel een verhoogd risico zijn op overlijden. Hartafwijkingen, hypocalciëmie en luchtwegmalacie (kraakbeendefecten lijden tot collaps van de luchtwegen) zijn risicofactoren voor een vroegtijdig overlijden op de kinderleeftijd. Het grootste deel van de kinderen groeit op tot volwassene.
Volwassenen kunnen ook vroegtijdig overlijden. De oorzaken hiervoor zijn veelzijdig en zijn met name gerelateerd aan cardiale defecten. Soms is sprake van een onverklaard plotseling overlijden.
Omdat er relatief weinig bekend is over volwassenen met 22q11.2DS en de oorzaken van hun onverklaard plotseling overlijden (dat soms achteraf voorkomen had kunnen worden), is het belangrijk om de medische problemen bij volwassenen met dit syndroom goed in kaart te brengen.
Naast het verkrijgen van multidisciplinaire zorg is ‘bijdragen aan kennis over volwassenen met 22q11.2DS één van de redenen om tenminste eenmalig een bezoek te brengen aan het expertisecentrum 22q11.2DS (Maastricht) of 's Heerenloo
Wist u dat......22q11.2DS een zeldzame aandoening is? Volgens de definitie vallen aandoeningen met een prevalentie van minder dan 1 op de 2.000 mensen onder de categorie 'zeldzame aandoeningen'.
Voor zeldzame aandoeningen zijn expertisecentra door de Nederlandse Federatie van Universitair Medisch Centra (NFU) landelijk opgericht. Waar u het expertisecentrum voor 22q11.2DS kunt vinden, leest u in het hoofdstuk 'Organisatie van de zorg'.